
PARA QUE SERVE A MODELAGEM COMPUTACIONAL DE PROTEÍNAS?
As proteínas expressam a informação genética e são formadas por uma sequência de aminoácidos. Além disso, a variação dessa sequência de aminoácidos formam diferentes tipos dessa molécula.
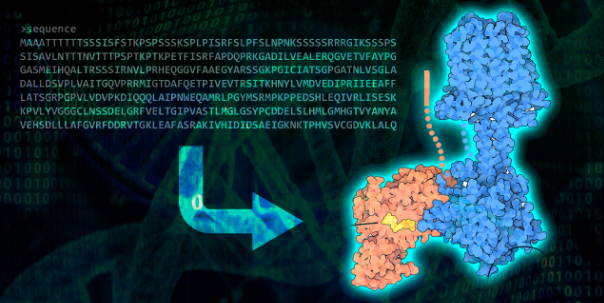
Com essa imensa diversidade de proteínas, é preciso de um banco de dados para conter todas essas informações. Por isso, usa-se a modelagem computacional que, por meio de softwares, modela a estrutura tridimensional das proteínas que não estão datadas. Essa técnica de modelagem computacional é classificada em dois tipos: baseadas em estruturas tridimensionais conhecidas e independentes de estruturas conhecidas. No primeiro grupo, a busca por uma nova proteína é menor que a segunda, porque já se sabe qual é a sua estrutura, e, precisa apenas modificá-la. Além disso, dentro desse caso, há uma divisão entre a modelagem comparativa (homóloga) e o threading. E, na metodologia independente de um molde, a busca é maior que a primeira, já que é necessário fragmentos ou a predição das proteínas secundárias (não referente a proteína desejada).
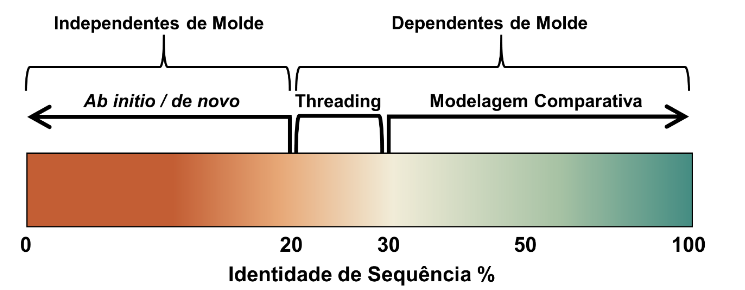
MODELAGEM COMPUTACIONAL DEPENDENTE DE MOLDE
A técnica de modelagem computacional dependente de molde se baseia no princípio de que as estruturas proteicas tendem a ficar conservadas ao longo do tempo, ou seja, ela considera que aconteça pequenas alterações nessas moléculas. Por isso, essa metodologia parte de estruturas pré-moldadas para modelar outra desconhecida.
MODELAGEM COMPARATIVA OU HOMÓLOGA
Como visto anteriormente, a modelagem comparativa (ou homóloga) é um tipo de técnica que se baseia em molde. Ela é bastante utilizada quando apenas se conhece os dados da sequência. Além disso, é necessário ter uma estrutura molde para usar essa técnica, então, para ter um modelo preciso, é preciso ter uma grande similaridade das sequências. Ademais, os servidores mais utilizados nesse método são: SWISS-MODEL e RaptorX.
THREADING
Já o threading, é utilizado quando a estrutura da proteína deseja e a sua similar (pré-moldada) tem baixa similaridade. Por isso, fragmenta a sequência para buscar por estruturas similares, diferente da técnica anterior, que procura a sequência inteira. Além disso, o software mais utilizado para modelar proteínas por essa técnica é o I-TASSER.
MODELAGEM COMPUTACIONAL INDEPENDENTE DE MOLDE
Como há uma imensa variedade de proteínas existentes, muitas delas têm pouca ou zero similaridade com as proteínas conhecidas, por isso, foi necessário o surgimento de uma técnica que não depende de molde. Essa técnica se baseia no pressuposto de que todas as proteínas se entrelaçam para um estado nativo ou para um grupo de estados que estejam no mínimo global de energia (menor nível de energia potencial). Além disso, existe uma metodologia, chamada de ab initio, que também é considerada como modelagem independente de molde.
AB INITIO
A metodologia ab initio se baseia puramente nas leis da Física. Esse método utiliza programas computacionais para prever os conhecimentos estruturais da proteína, como a previsão de ângulos de torção e inserção dos átomos por meio de cálculos matemáticos e estatísticos. Como essa técnica não se baseia em um molde, ela é mais lenta e não é recomendada em moléculas maiores que 200 aminoácidos. Além disso, pode-se utilizar dois softwares para a modelagem ab initio: QUARK e ROBETTA.
Deseja conferir mais conteúdos sobre o mundo da bioinformática e suas análises? Clique aqui e confira já em nosso blog!

REFERÊNCIAS
SILVA, L.; BASTOS, L.; SANTOS, L. Modelagem Computacional de Proteínas. Bioinfo, 2021. Disponível em: <bioinfo.com.br/modelagem-computacional-de-proteinas/>.
NEIS, A. Conceitos Básicos em Modelagem de Proteínas. Omixdata, 2019. Disponível em: <https://medium.com/omixdata/conceitos-b%C3%A1sicos-em-modelagem-de-prote%C3%ADnas-b9f8ac2c0b84>
NELSON, D. L.; COX, M. M. III. Aminoácidos, Peptídeos e Proteínas. Princípios de Bioquímica de Lehninger, v. 6, p. 75.