A compreensão das relações evolutivas entre seres vivos é um dos pilares da biologia moderna. A partir da proposição das teorias evolutivas por Charles Darwin em ‘’A origem das espécies’’ (1859), foi natural que houvesse a busca por ferramentas gráficas que representassem as relações do parentesco evolutivo entre espécies. Desse modo, traçam-se os diagramas das primeiras árvores filogenéticas, sugerindo diferentes hipóteses sobre a ancestralidade comum dos organismos.
Inicialmente, apenas as descrições morfológicas e comportamentais das espécies eram utilizadas como parâmetros de relação. Somado-se a essa imprecisão objetiva, os métodos de parcimônia eram amplamente empregados pela ausência de fundamentos teóricos mais sofisticados sobre as informações genéticas das espécies, ou seja, o caminho evolutivo mais simples, ou com menor número de alterações de caracteres, era preterido na elaboração das árvores filogenéticas. Tal método é exemplificado pela Figura 1, revelando que a hipótese evolutiva 1 seria mais parcimoniosa que a hipótese 2.
Figura 1: Exemplificação filogenia parcimoniosa
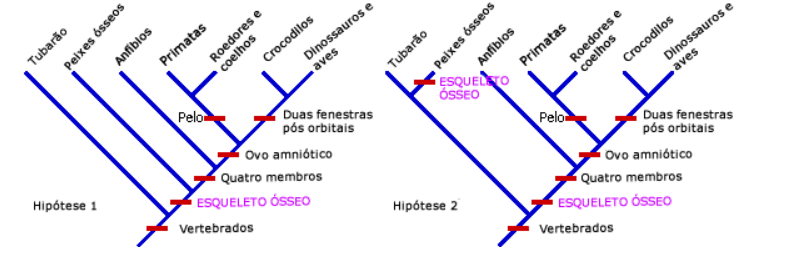
Fonte: Instituto de Biociências USP, [s.d].
Com o desenvolvimento das tecnologias de sequenciamento de ácidos nucleicos e do crescimento exponencial de dados genômicos, é fundamental a utilização de métodos computacionais para armazenar, processar, e, posteriormente, comparar e interpretar o conjunto de informações genéticas disponíveis. Nesse cenário, o campo de estudo da bioinformática integra o banco de dados ao tratamento estatístico com base em evidências moleculares para reconstruir as hipóteses evolutivas e padrões de ancestralidade mais complexos entre espécies. A árvore filogenética (Figura 2) evidencia um exemplo do emprego de métodos computacionais estatísticos para avaliar a proximidade evolutiva de diferentes espécies do gênero Bradypodion (camaleões africanos) a partir da comparação entre 15 genes selecionados.
Figura 2: Árvore filogenética para espécies do gênero Bradypodion
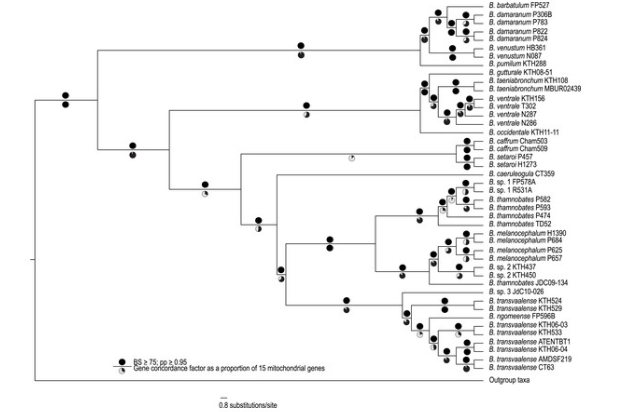
Fonte: MAIN et al (2024)
Os círculos pretos indicados acima das linha de cada clado representam as circunstâncias de duas medidas estatísticas utilizadas: BS (Bootstrap), o qual avalia a frequência percentual de ocorrência do agrupamento representado a partir de milhares de reamostragens testadas por programas computacionais, e pp (Posterior Probability), que avalia percentualmente a chance do agrupamento em questão estar correto a partir dos dados e do modelo empregados. Sendo assim, todos os clados que apresentam o círculo preto demonstraram robustez e confiabilidade estatística, tendo BS ≥ 75% e pp ≥ 95%.
Os círculos semi-preenchidos abaixo das linhas de cada clado representam o percentual de gCF (Gene Concordance Factor) encontrado, isto é, quantos dos 15 genes inicialmente identificados no ancestral comum geral sustentaram a criação de cada ramificação da árvore, fornecendo padrões evolutivos informativos que permitam a distinção de agrupamentos no cladograma. Círculos menos preenchidos revelam que poucos genes contribuíram para a formação do agrupamento em questão, sinalizando que há um conflito/ambiguidade de informações genéticas para tal clado (decorrentes, por exemplo, de hibridização de genes, convergência evolutiva, recombinação, taxas diferentes de mutação, etc.)
Ainda assim, com o acúmulo de informações genéticas com a disposição de ferramentas computacionais cada vez mais sofisticadas para tratar os dados genômicos, pode-se inferir, entre diversas aplicações, principalmente:
- trajetória evolutiva de espécies vivas e extintas;
- estudo da propagação e do combate de doenças para a epidemiologia molecular;
- biogeografia e padrões de dispersão;
- coevolução e relação entre organismos (hospedeiros e parasitas);
- sistemas de conservação para os táxons (grupos filogenéticos) de interesse com base na ancestralidade e nas informações evolutivas retratadas.
Com base nas árvores filogenéticas registradas nos bancos de dados genômicos, a Protos também oferece serviços de avaliação da funcionalidade de sequências genéticas de interesse. Para saber mais sobre nossos serviços, contate-nos!

REFERÊNCIAS:
FELSENSTEIN, Joseph. Evolutionary trees from DNA sequences: A maximum likelihood approach. Journal of Molecular Evolution, v. 17, n. 6, p. 368–376, 1981.
FELSENSTEIN, Joseph. Inferring phylogenies. 1° ed. Sunderland, Massachusetts: Sinauer Associates, 2004.
INSTITUTO DE BIOCIÊNCIAS USP. O que é parcimônia? EvoSite – Evolução 101, São Paulo, [s.d.]. Disponível em: https://ecologia.ib.usp.br/evosite/evo101/IIC1aUsingparsimony2.shtml. Acesso em 1 jul. 2025.
MAIN, Devon C. et al. The efficacy of single mitochondrial genes at reconciling the complete mitogenome phylogeny—a case study on dwarf chameleons. PeerJ, v. 12, 2024.
SILVA, Ruana Carolina Cabral da; ALVES, Maria Cidinaria Silva. O uso de ferramentas de bioinformática para análise de dados genéticos: uma revisão. Scientific Electronic Archives, v. 17, n. 1, 2024.